Thrust 1: machine-learned classical DFT and solvation models
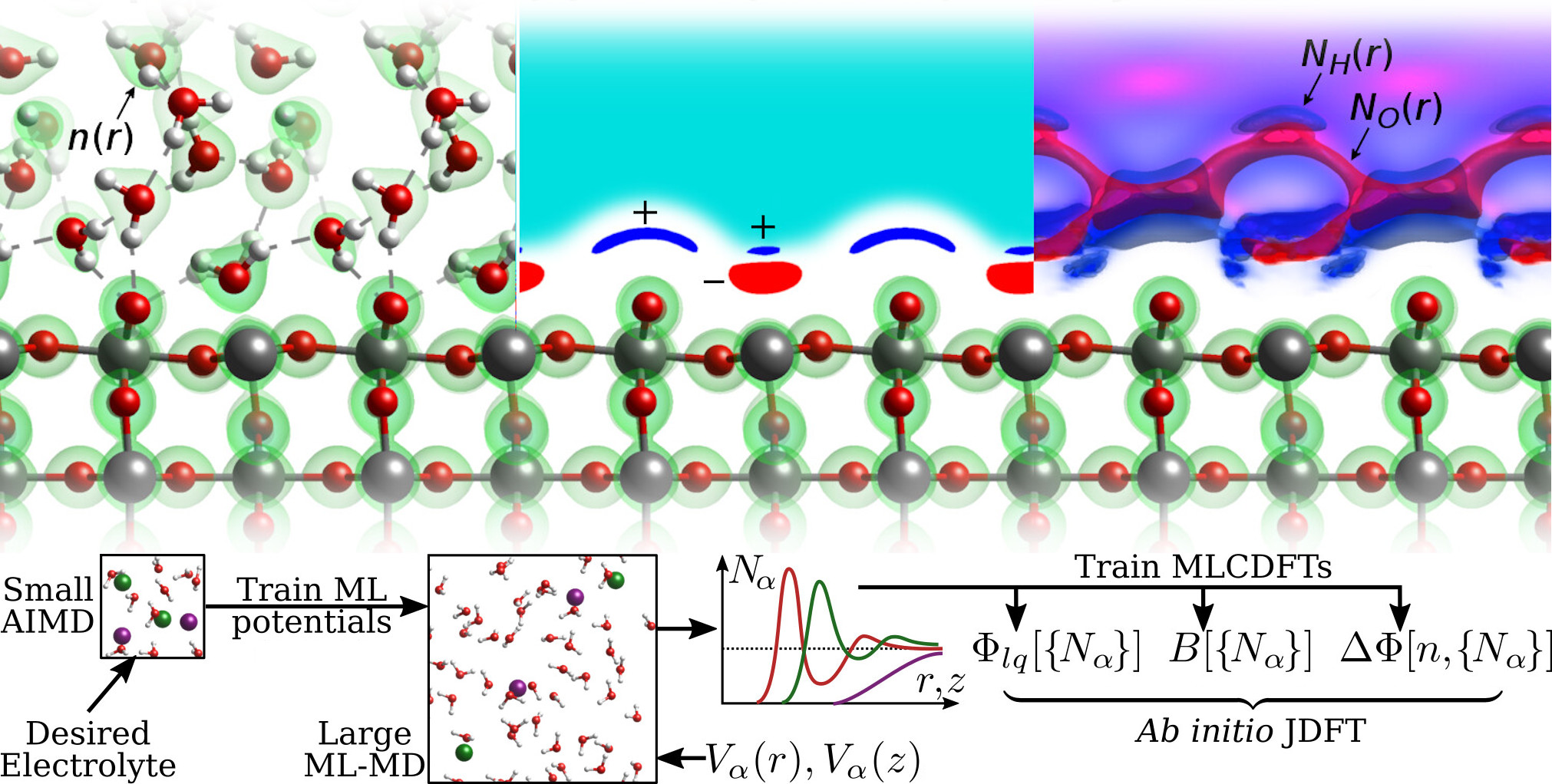
Accurate first-principles electrochemistry requires beyond-DFT methods to correctly capture the charge states of molecules on electrode surfaces, and accurate electrolyte solvation to determine the electrode charge at the specified potential within a grand-canonical electronic structure simulation. The expense of beyond-DFT methods such as RPA necessitate the avoidance of liquid/electrolyte thermodynamic sampling using molecular dynamics, which is possible using the framework of joint density-functional theory (JDFT). Advancing grand-canonical JDFT for first-principles electrochemistry requires techniques that can capture atomic-scale electrolyte structure in charged electrochemical interfaces.
Thrust 1 will develop the next generation of liquid density models that capture the electrochemical double layer structure for accurate first-principles electrochemical calculation using grand-canonical JDFT methods. Specifically, we will develop free energy functionals for classical DFT and bridge functionals for RISM treatment of arbitrary electrolytes, as well as electrode-electrolyte coupling functionals, all using a new machine learned classical DFT approach. We will train these models exclusively to ab initio data, using large molecular dynamics simulations using ML potentials trained to small AIMD simulations, making it possible to automatically develop ab initio electrochemical solvation for any electrolyte.