GC-DFT predictions of CO2R on Cu and Ag published in J. Phys. Chem. C
Y. A. Alsunni, A. W. Alherz, and C. B. Musgrave, “Electrocatalytic Reduction of CO2 to CO over Ag(110) and Cu(211) Modeled by Grand-Canonical Density Functional Theory”, J. Phys. Chem. C 125, 23773 (2021)
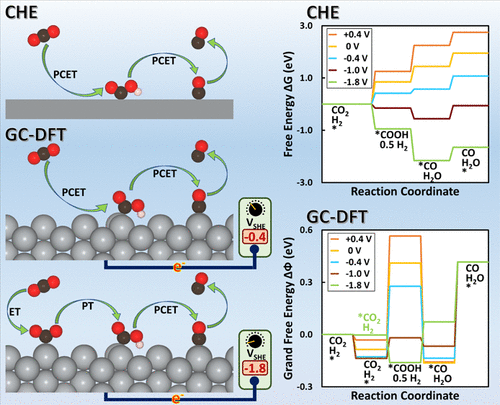
We report the results of modeling CO2 reduction (CO2R) to CO over Ag(110) and Cu(211) surfaces at different applied potentials using grand-canonical density functional theory (GC-DFT), a method specifically designed to accurately model electrochemical systems. In addition to demonstrating GC-DFT’s ability to accurately model electrochemical processes, we also compare it with the computational hydrogen electrode (CHE) approach. GC-DFT predicts that the geometries of these reacting systems strongly depend on the applied potential, and the Helmholtz free energies vary nonlinearly with the applied potential, which contradicts a central assumption of the CHE approach. The CHE approach neglects the change in the number of electrons on the electrode surface at different applied potentials, which reduces its accuracy as the potential changes from the potential of zero charge. Our results further demonstrate that the grand free energies of the reaction intermediates not only depend on the value of the applied potential but also on the metal surface type, adsorption site, and adsorbate. GC-DFT’s ability to predict the effect of the applied potential on adsorbate geometry enables it to evaluate different possible reaction mechanisms at different applied potentials. For instance, GC-DFT predicts that the first step of CO2R likely switches from proton-coupled electron transfer to sequential electron transfer and then proton transfer at more reducing potentials, a result that cannot be determined by the CHE because it assumes that all electron transfers are coupled to proton transfers and neglects the effect of the applied potential on the adsorbate geometry.